ConSurf Gallery
Click on the linked example titles to view ConSurf results and the rotatable molecules colored according to ConSurf conservations scores, or click on the static figures to see them in greater detail.

Color coding scheme of ConSurf
Example 1: MHC Class I Heavy Chain (H-2Kb, PDB ID 2VAA chain A).
Complexed With Beta-2 Microglobulin (Chain B) and
Vesicular Stomatitis Virus Nucleoprotein (fragment 52-59, chain P).
Here, both variability and conservation reflect
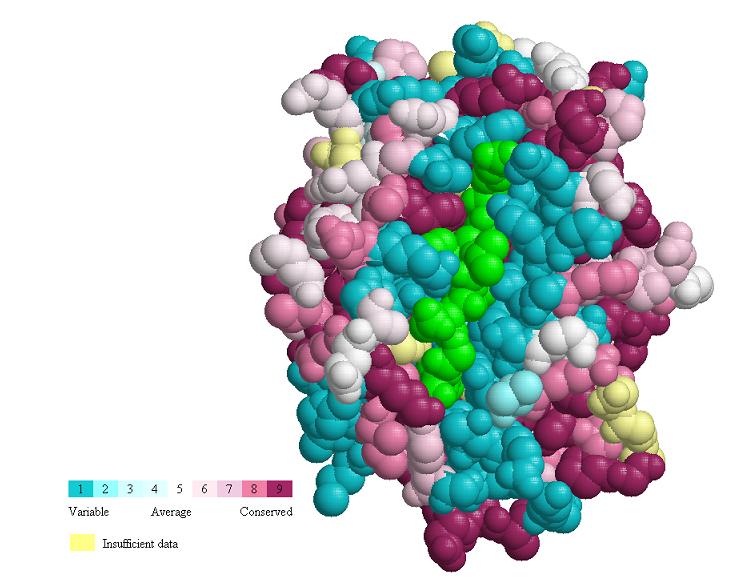
function. Functional variability is seen in the peptide-binding groove
(Fig. MHC_1, accomodating chain P). Allelic diversity in the population,
concentrated in the inner surface of the groove, enables the population
to present the maximal range of foreign (e.g. viral) peptides to T lymphocytes,
for defensive immune responses. Conserved patches suggest functions, some
of which may not yet be identified. Gln226 is known to be important in
binding the CD8 coreceptor of the T cell (Kern
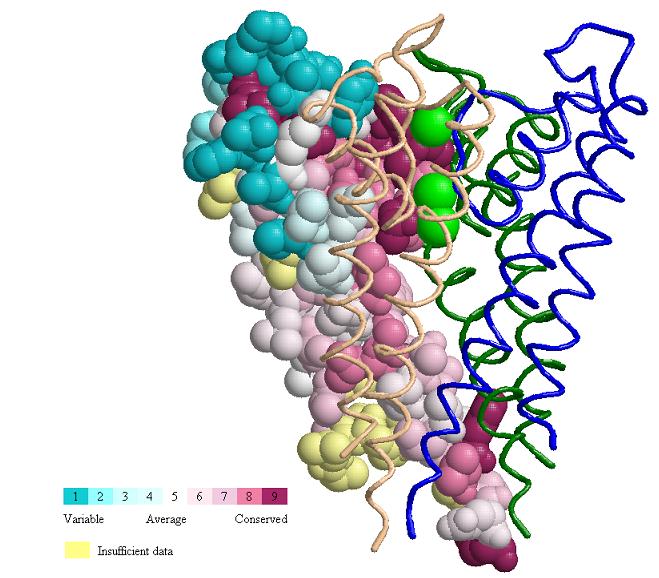
et al.). Conservation is also evident in the interface binding chain
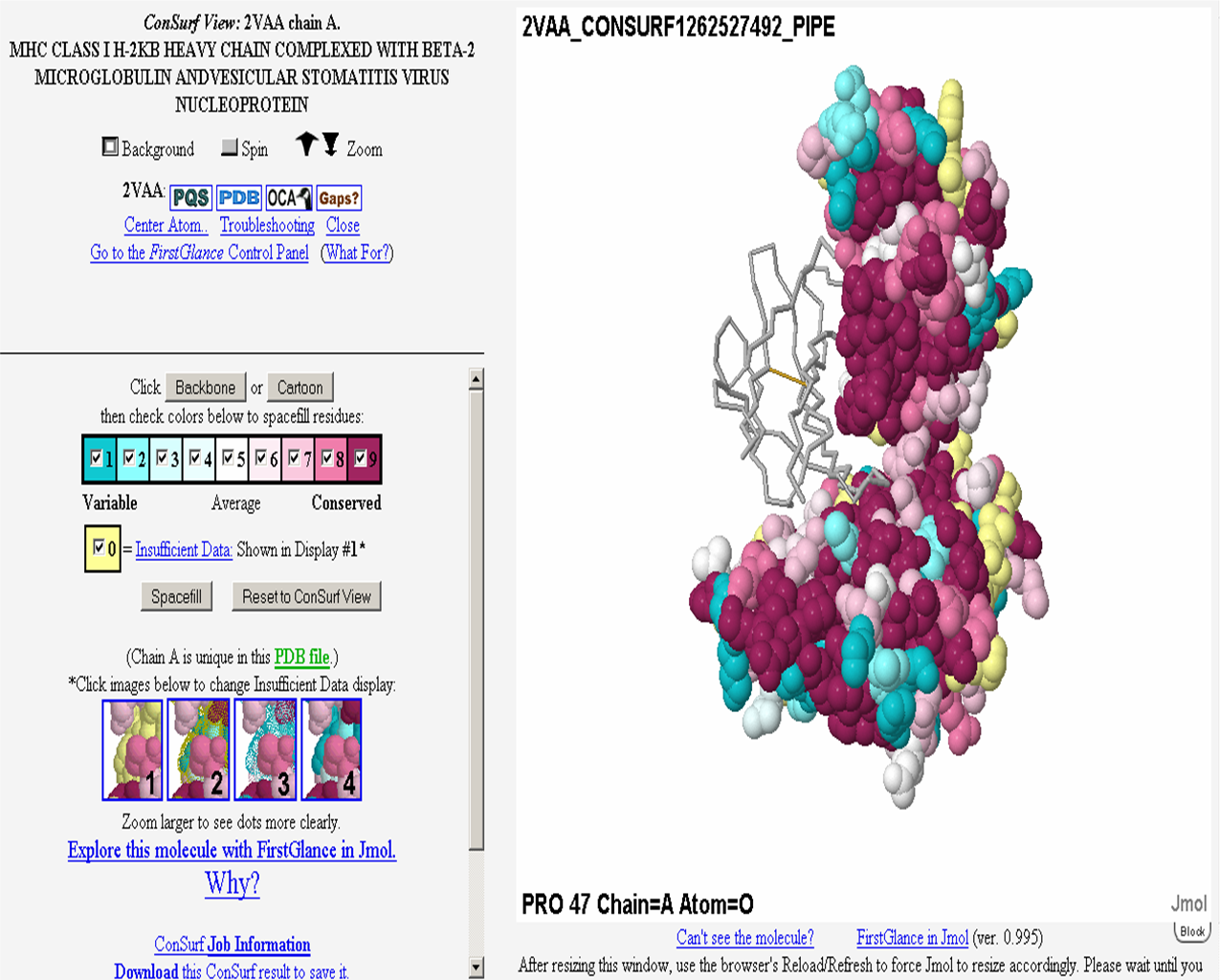
B (Fig. MHC_2). Fig. MHC_3 shows a general view of the Protein explorer
graphic page.
More on MHC structure is at the
Lehninger
and Martz
sites.
 |
 |
 |
| Fig. MHC_1 | Fig. MHC_2 | Fig. MHC_3 |
Example 2: Potassium
Channel (Kcsa, PDB ID 1bl8 chain A)
Potassium Channel (Kcsa, chains: A, B, C, D) From
Streptomyces Lividans
The potassium channel is an integral membrane protein
with sequence similarity to all known K+ channels, particularly in the
pore region. In Fig. 1bl8_1, we can see a general view of the conservation
pattern of one of the four identical subunits (chain A, all other subunits
in strands, K+ atoms yellow), which shows that ConSurf clearly detects
the conservation among the amino acids facing the pore region (critical
K+ signature sequence aa). Fig. 1bl8_2, shows in greater detail the the
contact between a K+ atom and the highly conserved G77, Y78 and G79 amino
acids, which are known to be absolutely required for K+ selectivity.
More on the the structure and function of the potassium
channel at (Doyle
et al.)
 |
 |
| Fig. 1bl8_1 | Fig. 1bl8_2 |
Example 3: Human Insulin (protein sequence and structure analysis) (INS_HUMAN)
Proinsulin is the prohormone precursor to insulin made in the beta cell of the islets of Langerhans. In humans,
proinsulin is encoded by the INS gene. Proinsulin is processed by a series of proteases to form mature insulin. During this process
the Signal peptide (1-24) and the C peptide (57-87) are removed. The remaining peptides (Insulin A Chain and Insulin B chain) are
connected by disulfide bonds and constitute the mature insuline.
In figure INS_HUMAN (located below) the ConSurf analysis for INS_HUMAN is shown. The figure reveal that the Insulin B (25-54)
and Insulin A (90-110) peptides are highly conserved. In addition the disulfide bonds (CYS31-CYS96, CYS43-CYS109 and CYS95-CYS100)
which are crucial to the structure of the mature insuline are also highly conserved compared to the removed peptides.
More about the human proinsulin at (Nicol et al),
(Oyer et al.) and
(Ko et al).
In addition using the ConSurf result page the user can also review the projection of the
conservation scores calculated for the sequence onto the insulin structures.
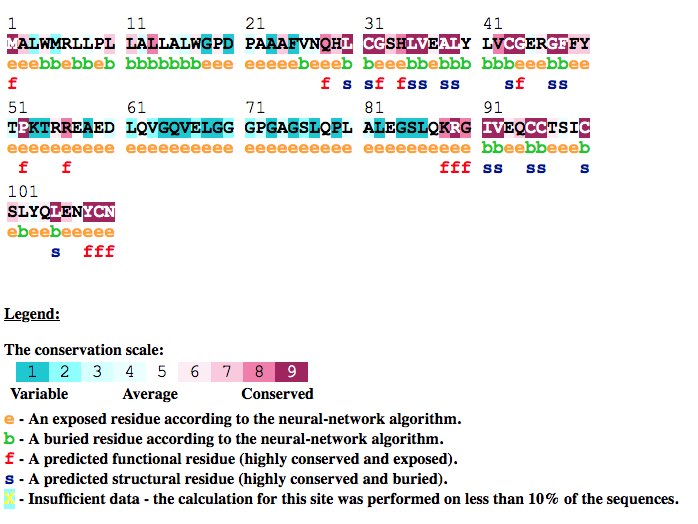
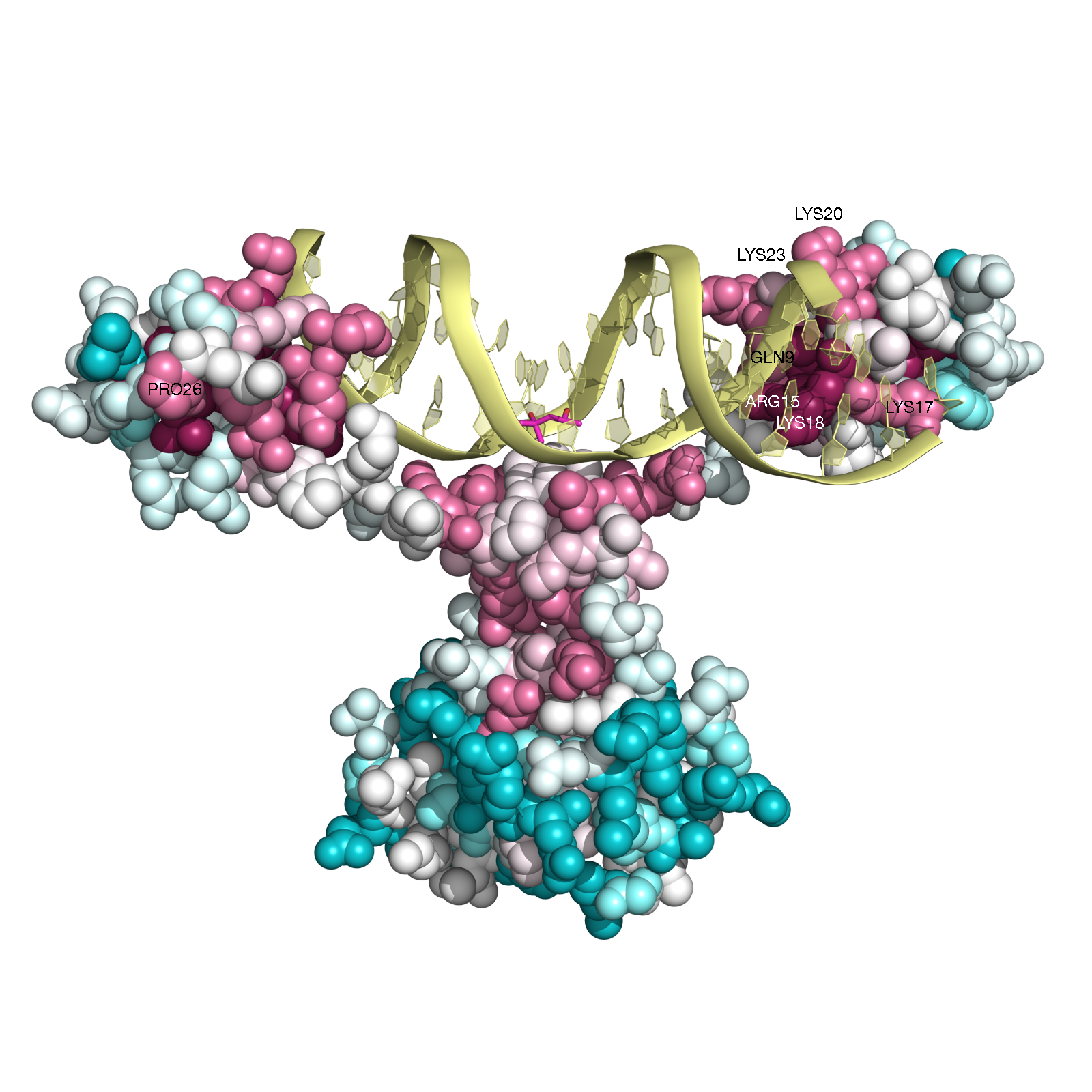
Example 4: Gal4 transcription factor in yeast (PDB ID 3coq chain A)
Gal4 is a transcription factor in yesat. This protein is a positive regulator for the gene expression of the galactose-induced genes such as GAL1, GAL2, GAL7, GAL10, and MEL1 which encode for the enzymes used to convert galactose to glucose. It recognizes a 17 base pair sequence in (5'-CGGRNNRCYNYNCNCCG-3') the upstream activating sequence (UAS-G) of these genes.
ConSurf analysis (Figure GAL4, below) reveals, as expected, that the functional
regions of this protein are highly conserved. For example, all the cysteins that form the Zn(2)-C6 DNA binding domain (CYS11, CYS14, CYS21,
CYS28, CYS31, CYS38) (Pan and Coleman, 1990) receive the highest
conservation scores. Likewise, PRO26, that is known to be central for DNA binding
(Johnston, 1987) is also highly conserved according to our analysis.
In addition, other amino-acids residues, which are in contact with the DNA (i.e., GLN9, LYS17, LYS18, LYS20, ARG15, LYS23)
(Marmorstein et. al. 1992) are also relatively conserved.
ConSurf was also applied to nucleic acid sequences from yeast, which are the known binding sites of GAL4 and their adjacent neighborhood. As anticipated, the analysis reveled that the consensus pattern CGG-N11-CCG typical to GAL4 binding site is highly conserved (see ConSurf full results for the nucleic acid sequences here)
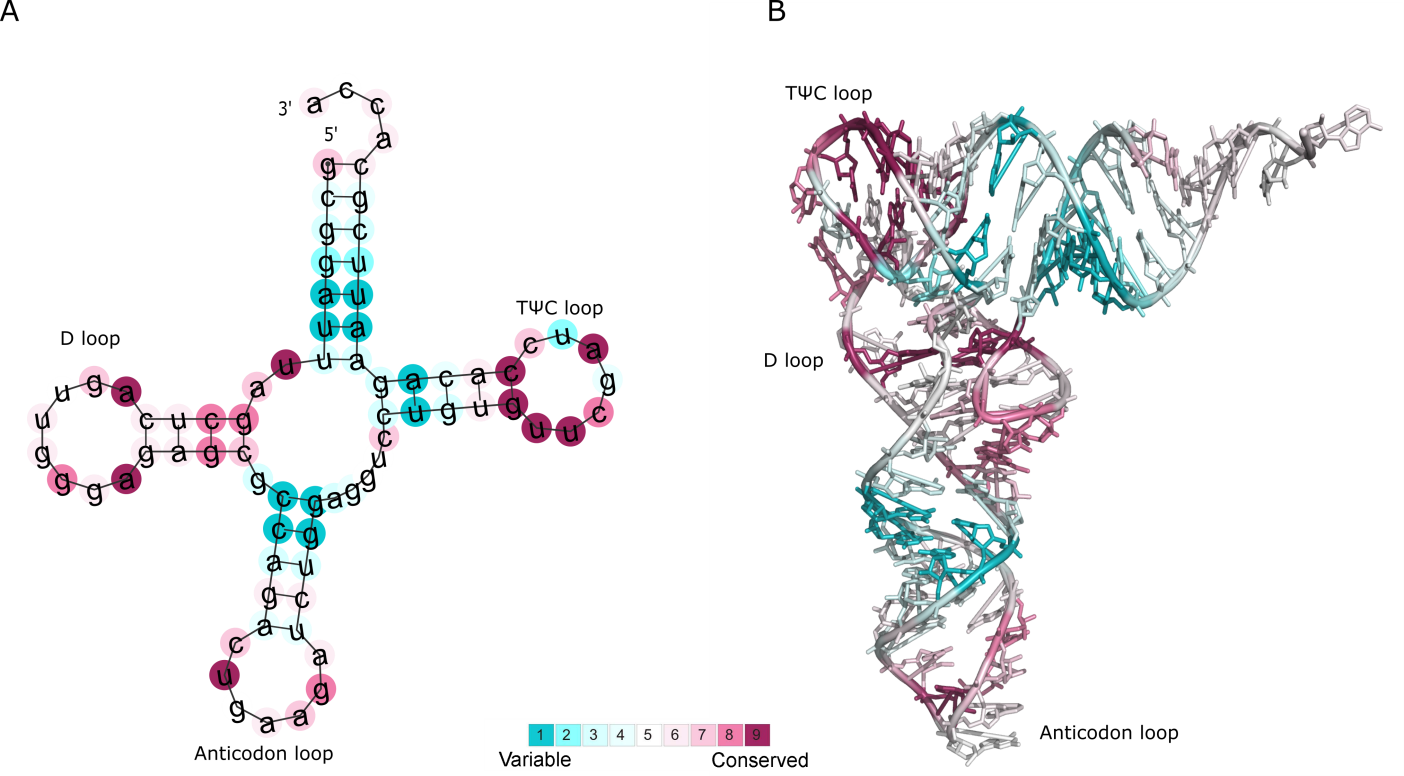
Example 5: Phe-tRNA.
For RNA sequence queries, ConSurf offers the possibility to predict the secondary structure. Structures are predicted using
the RNAfold program of the Vienna package (Lorenz,R et. al. 2011,
Mathews,D.H. et. al. 2004), and the structure with the lowest free
energy is selected. The ConSurf conservation grades are mapped onto the predicted secondary structure. Correlating the evolutionary data with
the structural model offers the means to quickly detect functional regions within the RNA query. To exemplify this feature, we analyze the
well-studied Phe-tRNA molecule from yeast (Figure Phe-tRNA A).
The calculation is based on RFAM homologous sequences
(Nawrocki,E.P. et. al. 2015) of the Phe-tRNA molecule
(RFAM RF00005 family) clustered by CD-HIT
(Li W and Godzik A. 2006) to the level of 80% sequence identity and
aligned using MAFFT (Katoh K and Standley DM. 2013).
The results show that some bases in the TΨC and D loops are assigned with particularly
high conservation grades (colored in magenta). Some of these positions are known to be of structural and functional importance
(Zagryadskaya E.I. et. al 2003,
Guy,M.P. et. al. 2014).
Figure Phe-tRNA B demonstrates the projection of ConSurf grades onto the 3D structure of the Phe-tRNA molecule (PDB id: 1EHZ chain A), and
further highlights the importance of the evolutionarily conserved positions.
Example 6: β subunit of DNA polymerase III from Escherichia coli.
The new version of ConSurf automatically suggests the possibility to map the calculated evolutionary conservation grades of
the amino acids not only onto the single asymmetric unit chain, which is often deposited in the PDB, but also on the protein assembly as predicted
by PISA (http://www.ebi.ac.uk/pdbe/prot_int/pistart.html).
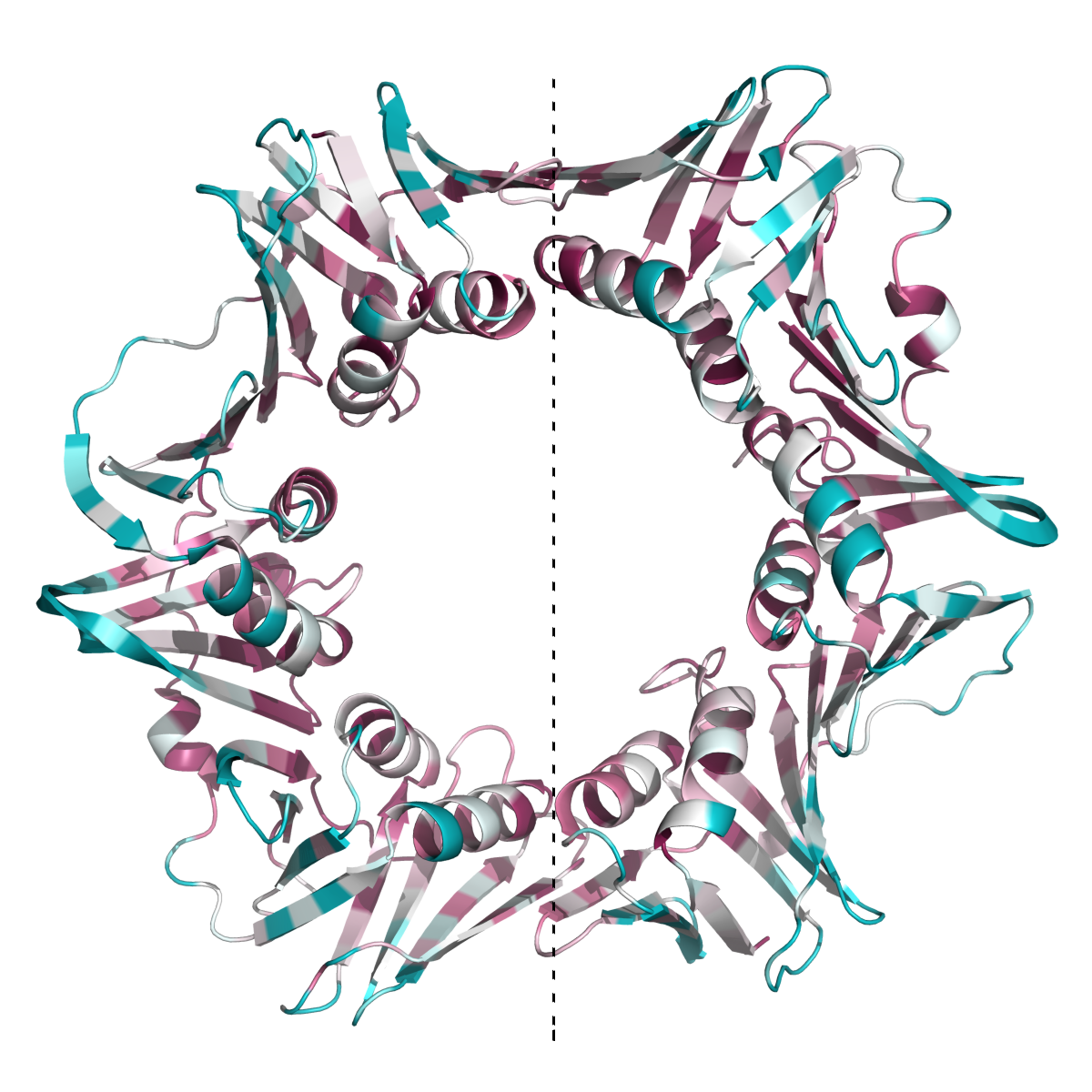
This new feature is demonstrated by analyzing the 3D structure of the β subunit of DNA polymerase III from Escherichia coli (Figure 2POL).
This protein functions as homo-dimer (PDB ID: 2POL),
and as anticipated, most of the residues at the inter-subunit interfaces
(Kong, X.P. et. al. 1992) are highly evolutionarily conserved
(Leu108, Lys74, Ile272, Leu273, Glu300, Glu304). The interfaces between the two subunits of the homodimer (on both sides of the dotted line)
are highly conserved, as well as the internal face of the ring, which interacts with the DNA.