THE CONSURF SERVER
SERVER FOR THE IDENTIFICATION OF FUNCTIONAL REGIONS IN PROTEINS
Table of contents
- Introduction
- Conservation scores
- Methodology
- Confidence interval of the inferred conservation scores
- Advanced Options and Detail
- Coloring Scheme
- PDB 3D structure
- Dealing with unreliable positions
- Searching for homologous sequences
- Output
- Generating the phylogenetic tree
- Example
- Calculating the amino acid conservation score
- ConSurf vs estimates of the evolutionary conservation without explicit use of phylogenetic relationship
- Analyzing nucleic acid sequences
- References
- Model of substitution for protein
Introduction
The ConSurf server (Glaser et al., 2003; Landau et al., 2005; Ashkenazy et al., 2010; Celniker et al., 2013; Ashkenazy et al., 2016) is a bioinformatics tool for estimating the evolutionary conservation of amino/nucleic acid positions in a protein/DNA/RNA molecule based on the phylogenetic relations between homologous sequences. The degree to which an amino (or nucleic) acid position is evolutionarily conserved (i.e., its evolutionary rate) is strongly dependent on its structural and functional importance. Thus, conservation analysis of positions among members from the same family can often reveal the importance of each position for the protein (or nucleic acid)'s structure or function. In ConSurf, the evolutionary rate is estimated based on the evolutionary relatedness between the protein (DNA/RNA) and its homologues and considering the similarity between amino (nucleic) acids as reflected in the substitutions matrix (Pupko et al., 2002; Mayrose et al., 2004). One of the advantages of ConSurf in comparison to other methods is the accurate computation of the evolutionary rate by using either an empirical Bayesian method (Mayrose et al., 2004) or a maximum likelihood (ML) method (Pupko et al., 2002).
Methodology
Given the amino or nucleic acid sequence (can be extracted from the 3D structure), ConSurf carries out a search for close homologous sequences using MMsecs2, HMMER, CSI-BLAST, PSI-BLAST or BLAST (Finn et al., 2011; Altschul et al., 1990, 1997; Biegert and Söding, 2009). The user may select one of several databases and specify criteria for defining homologues. The user may also select the desired homologues from the hits. The sequences are clustered and highly similar sequences are removed using CD-HIT (Li and Godzik, 2006). A multiple sequence alignment (MSA) of the homologous sequences is constructed using MAFFT (default), PRANK, T-COFFEE, MUSCLE or CLUSTALW. The MSA is then used to build a phylogenetic tree using the neighbor-joining algorithm as implemented in the Rate4Site program (Pupko et al., 2002). Position-specific conservation scores are computed using the empirical Bayesian or ML algorithms (Mayrose et al., 2004; Pupko et al., 2002). The continuous conservation scores are divided into a discrete scale of nine grades for visualization, from the most variable positions (grade 1) colored turquoise, through intermediately conserved positions (grade 5) colored white, to the most conserved positions (grade 9) colored maroon. The conservation scores are projected onto the protein/nucleotide sequence and on the MSA (A flowchart of ConSurf is shown in Fig 1 and detailed below)
(Optional)
(Optional)
Sequence
(Optional)
Figure 1: A flowchart of ConSurf pipeline.
Advanced Options and Details
PDB 3D structure
Three-dimensional structures of biological macromolecules are available in the Protein Data Bank (PDB)(Rose et al., 2015), a copy of which is accessible from the ConSurf server. AlphaFold model structures of most other proteins are available in UniProt. The HMMER, CSI-BLAST or PSI-BLAST (Finn et al., 2011; Biegert and Söding, 2009; Altschul et al., 1997, 1990) search for homologues is done using the target chain sequence extracted from the SEQRES record of the PDB file as the input query. If the SEQRES record does not exist, ConSurf extracts the sequence from the ATOM record. Thus, a user-provided PDB file must include the ATOM record.
Searching for homologous sequences
For proteins, the server uses the MMseqs2, HMMER, CSI-BLAST or PSI-BLAST (Finn et al., 2011; Altschul et al., 1997; Biegert and Söding, 2009; Steinegger and Söding, 2017) heuristic algorithm with default parameters to collect homologous sequences of a single polypeptide chain of known 3D-structure. The search can be carried out using the following databases:
- UniRef90 - consists of cluster sequences and sub-fragments with 11 or more residues that have at least 90% sequence identity with each other (from any organism) into a single UniRef entry, displaying the sequence of a representative.
- SWISS-PROT - a curated protein sequence database which strives to provide a high level of annotation.
- UniProt - the universal protein resource, a central repository of protein data created by combining SWISS-PROT, TrEMBL and PIR.
- Clean UniProt - a modified version of the UniProt database aimed to screen the more reliable sequences using two criteria:
- a. If the "Decription" (DE) field contain "Disease", "RIKEN", "variant", "mutation", "mutant" or "whole genome shotgun sequence" the sequence is removed;
- b. If the database is "TrEMBL" and the "Comments" (CC) lines contain the word "CAUTION" the sequence is removed.
- NR database - 'non-redundant' database (i.e. with duplicated sequences removed). NR contains non-redundant sequences from GenBank together with sequences from other databanks (Refseq, PDB, SWISS-PROT, PIR and PRF).
The user can also set the maximum number of homologs to collect and the number of iterations while the default runs one HMMER iteration, with maximum of 150 homologs and E-value cutoff of 0.0001 (E-value describes the number of hits one can "expect" to see just by chance when searching a database of a particular size. The higher the E-value, the more hits will be expected, but the pairwise distance between them and the query sequence will increase). The user can also control the maximal percentage identity between sequences, removing redundant sequences and specify the level of redundant sequences for removal. The sequences found are clustered by their level of identity using CD-HIT (Li and Godzik, 2006) and the cutoff specified by the user (default level is 95% identity). The minimal percentage for homologs is set by default to 35% which is the level of the upper bound of the 'twilight zone' for protein structures (Rost, 1999). See figure 2 for user's MSA parameters screen on the web server.
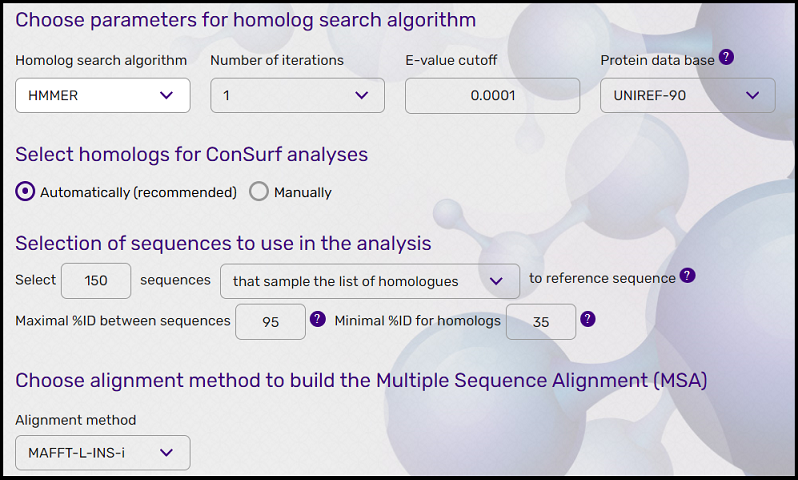
Figure 2: Screenshot of the parameters window to build the Multiple Sequence Alignment (MSA) on the Consurf web server.
Note: The best performance is obtained when single domains are used. Using a polypeptide chain that contains several domains often results in finding proteins that are homologous to one of the domains but not to the others. Therefore it is advisable to analyze multi-domain proteins one domain at a time.
For nucleic acid sequence the search is performed using the HMMER/BLAST algorithm (Biegert and Söding, 2009; Altschul et al., 1990) on the NCBI NT database. The default runs one BLAST iteration with E-value cutoff of 0.001.
Generating the phylogenetic tree
The server constructs a phylogenetic tree that is consistent with the available MSA, using the neighbor joining (NJ) algorithm (Saitou and Nei, 1987), as implemented in the Rate4Site program (Pupko et al., 2002). The server can also accept a user-provided phylogenetic tree (in Newick format). This option can be used only if a corresponding MSA is provided.
Calculating the conservation scores
The conservation score at a site corresponds to the site's evolutionary rate. The rate of evolution is not constant among amino (nucleic) acid sites: some positions evolve slowly and are commonly referred to as "conserved", while others evolve rapidly and are referred to as "variable". The rate variations correspond to different levels of purifying selection acting on these sites. For example, in proteins, the purifying selection can be the result of geometrical constraints on the folding of the protein into its 3D structure, constraints at amino acid sites involved in enzymatic activity or in ligand binding or, alternatively, at amino acid sites that take part in protein-protein interactions.
In ConSurf, the rate of evolution at each site is calculated using either the empirical Bayesian (Mayrose et al., 2004) or the Maximum Likelihood (Pupko et al., 2002) paradigm. In both of these methods, the stochastic process underlying the sequence evolution and the phylogenetic tree are explicitly taken into account. The Bayesian method was shown to significantly improve the accuracy of conservation scores estimations over the Maximum Likelihood method, in particular when a small number of sequences are used for the calculations (Mayrose et al., 2004). An additional advantage of the Bayesian method is that a confidence interval is assigned to each of the inferred evolutionary conservation score.
Analyzing nucleic acid sequences
Despite increasing interest in the non-coding fraction of transcriptomes, the number, the level of conservation, and functions, if any, of many non-protein-coding transcripts remain to be discovered. However, it has already been shown that many of the non-coding sequences are connected to regulatory processes. ConSurf offers estimations of the evolutionary rate for each position of nucleic acid sequences in the same manner used for amino acid residues. For that purpose, four evolutionary models were implemented in the Rate4Site program: (i) the Juke and Cantor 69 model (JC69), which assumes equal base frequencies and equal substitution rates (Jukes and Cantor, 1969). (ii) The Tamura 92 model that uses only one parameter, which captures variation in G-C content (Tamura, 1992). (iii) The HKY85 model, which distinguishes between transitions and transversions and allows unequal base frequencies (Hasegawa et al., 1985). (iv) The General Time Reversible (GTR) model, which is the most general time-reversible model. The GTR parameters consist of an equilibrium base frequency vector, giving the frequency at which each base occurs at each site, and the rate matrix (Tavare, 1986). When enough data (i.e. sequences) are available, the GTR model is superior over the more simplified Tamura 92 model. However, the Tamura 92 model is recommended in cases in which the data are not sufficient for reliable estimation of the model parameters and thus it is the default option for analyzing nucleic acid sequences in ConSurf. ConSurf also allows automatic selection of the model that best fits the analyzed sequences, as determined by the Akaike information criterion (AIC) (Akaike, 1974; Darriba et al., 2012, 2011).
Model of substitution for proteins
The inference of evolutionary conservation relies on a specified probabilistic model of amino-acid replacements (Pupko et al., 2002; Mayrose et al., 2004). The server supports a few models of substitution for nuclear DNA-encoded proteins as well as models of non-nuclear DNA-encoded proteins. The model of substitution can be chosen from the "Evolutionary Substitution Model" drop-down list. The LG (Le and Gascuel, 2008), JTT (Jones et al., 1992), Dayhoff (M.O. Dayhoff, R.M. Schwartz, and B. C, 1978) and WAG (Whelan and Goldman, 2001) matrices are suited for nuclear DNA-encoded proteins. The WAG matrix has been inferred from a large database of sequences comprising a broad range of protein families and is thus suited for distantly related amino acid sequences (Whelan and Goldman, 2001). The LG matrix (Le and Gascuel, 2008), incorporates variability of evolutionary rates across sites in the matrix, and was shown to outperform other substitutions matrices for proteins. The mtREV (Adachi and Hasegawa, 1996) and cpREV (Adachi et al., 2000) matrices are suitable for mitochondrial, and chloroplast DNA-encoded proteins, respectively. As in the case of analyzing nucleic acid sequences, the model that best fits the analyzed sequences can be automatically selected by ConSurf.
Conservation Scores
The conservation scores calculated by ConSurf appear in the SCORE column in the "Per-Residue Details" link in the outout page. The scores are normalized, so that the average score for all residues is zero, and the standard deviation is one. The conservation scores calculated by ConSurf are a relative measure of evolutionary conservation at each sequence site of the target chain. The lowest score represents the most conserved position in a protein (DNA/RNA). It does not necessarily indicate 100% conservation (e.g. no mutations at all), but rather indicates that this position is the most conserved in this specific protein (DNA/RNA) calculated using a specific MSA.
Confidence interval of the inferred conservation scores
Positions in the MSA exhibiting too little variation caused by too few sequences or too little diversity among sequence homologs can render evolutionary analysis meaningless (Thornton et al., 2000). Using the Bayesian method to calculate evolutionary conservation, confidence intervals for the conservation scores estimations are obtained (Susko et al., 2002). When the number of sequences is small, the confidence interval tends to be large, meaning a low level of support to the inferred conservation score. When the number of sequences increases, confidence intervals become smaller, and the point score estimates are more assured. In ConSurf, a confidence interval is assigned to each of the inferred evolutionary conservation scores. The confidence interval is defined by the lower and upper quartiles (the 25th and 75th percentiles of the inferred evolutionary rate distribution, respectively). This measure gives the 50% confidence interval and also indicates on the dispersion of each of the estimated scores. Amino acid positions that are assigned confidence intervals that are too large to be trustworthy are marked as having insufficient data in the output files of the server.
Coloring Scheme
The continuous conservation scores are partitioned into a discrete scale of 9 bins for visualization, such that bin 9 contains the most conserved positions and bin 1 contains the most variable positions. The color grades (1-9) are assigned as follows: The conservation scores below the average (negative values, which are indicative of slowly evolving, conserved sites) are divided into 4.5 equal intervals. The same 4.5 intervals are used for the scores above the average (positive values, which are indicative of rapidly evolving, variable sites). Thus, 9 equally sized categories of conservation are obtained. However, because the conservation distribution is asymmetrical around the average, the range of grade 1 is extended to include the most variable grades. Namely, the range of grade 1 is larger than the rest. Colors are then assigned to the 9 grades for graphic visualization. The width of each color grade varies for different polypeptide chains using this procedure. That is, the coloring results of a ConSurf run do not indicate the absolute magnitudes of evolutionary distances, but rather the relative degree of conservation of each amino acid position. ConSurf scaling procedure does not guarantee that grades 1-8 will always be occupied, although grade 9 is always occupied by at least one residue.
Dealing with unreliable positions
Conservation scores that are obtained for positions in the alignment that have less than 6 un-gapped amino acids are considered to be unreliable. When using the Bayesian method (Mayrose et al., 2004) for the conservation scores calculations, confidence intervals around the estimated rates are computed. The high and low values of each interval are assigned color grades according to the 1-9 coloring scheme. If the interval in a specific position spans 4 or more color grades the score is considered as unreliable. Such positions are colored light yellow in the graphic visualization output.
Output
If a 3D structure of the protein (or of the DNA/RNA) is provided:
- The nine-color conservation scores are projected onto the 3D structure of the query protein and the colored protein structure is shown by FirstGlance in Jmol or Mol*.
- Scripts for visualizing the protein colored with ConSurf scores are generated for PyMol; (DeLano, 2008), UCSF-ChimeraX (Pettersen et al., 2021), Jmol; (Herráez, 2006).
For proteins, ConSurf also automatically suggests the possibility to map the calculated evolutionary conservation grades of the amino acids not only onto the single asymmetric unit chain, which is often deposited in the PDB, but also on the protein assembly as predicted by PISA
For proteins in which the 3D structure was not provided by the user, an up-to-date version of the Protein Data Bank (Rose et al., 2015) is searched for relevant homologues. If a structure of at least one homologous protein is available, the user may map the conservation scores on the structure. This option should ease the procedure for the non-expert users, who may be unfamiliar with the 3D structure. In addition, ConSurf goes one step further and uses HHPred (Meier and Söding, 2015) and MODELLER (Sali and Blundell, 1993) to produce a homology-model of the query (using default parameters). Briefly, HHPred uses a hidden Markov model (HMM) to search for potential templates of known 3D structure in the PDB (Remmert et al., 2012), and the MODELLER algorithm (Sali and Blundell, 1993) uses them to predict a 3D model for the query sequence. The ConSurf conservation grades are then mapped onto the predicted model.
For RNA sequence queries, ConSurf offers the possibility to predict the secondary structure. Structures are predicted using the RNAfold program of the Vienna package (Lorenz et al., 2011; Mathews et al., 2004), and the structure with the lowest free energy is selected. The ConSurf conservation grades are mapped onto the predicted secondary structure. Correlating the evolutionary data with the structural model offers the means to quickly detect functional regions within the RNA query.
For all cases, ConSurf creates the following outputs:
- The sequence and MSA colored by ConSurf conservation scores.
- A text file that summarizes for each position the normalized score calculated, the assigned grade (1-through-9), the reliability estimation (for the Bayesian method) and the amino acids/nucleotides observed in the respective MSA column.
- The sequences selected for the MSA and the MSA constructed (unless those files were uploaded by the user).
- A file with the frequency of each amino acid/nucleotide observed in each column of the MSA.
- The evolutionary tree, which was calculated by the server or uploaded by the user, together with the MSA are shown using the WASABI platform (Veidenberg et al., 2015). Moreover, using WASABI, a user can select a subtree containing a fraction of the homologous sequences and conduct a follow-up ConSurf analysis with these selected sequences. To refine a ConSurf analysis to a selected subtree, a user can choose any internal node on the phylogenetic tree and open a WASABI menu by using a right mouse click. Selecting the option 'run ConSurf on subtree' will issue a new window with the new ConSurf run for the selected subtree sequences (see example in Figure 3).
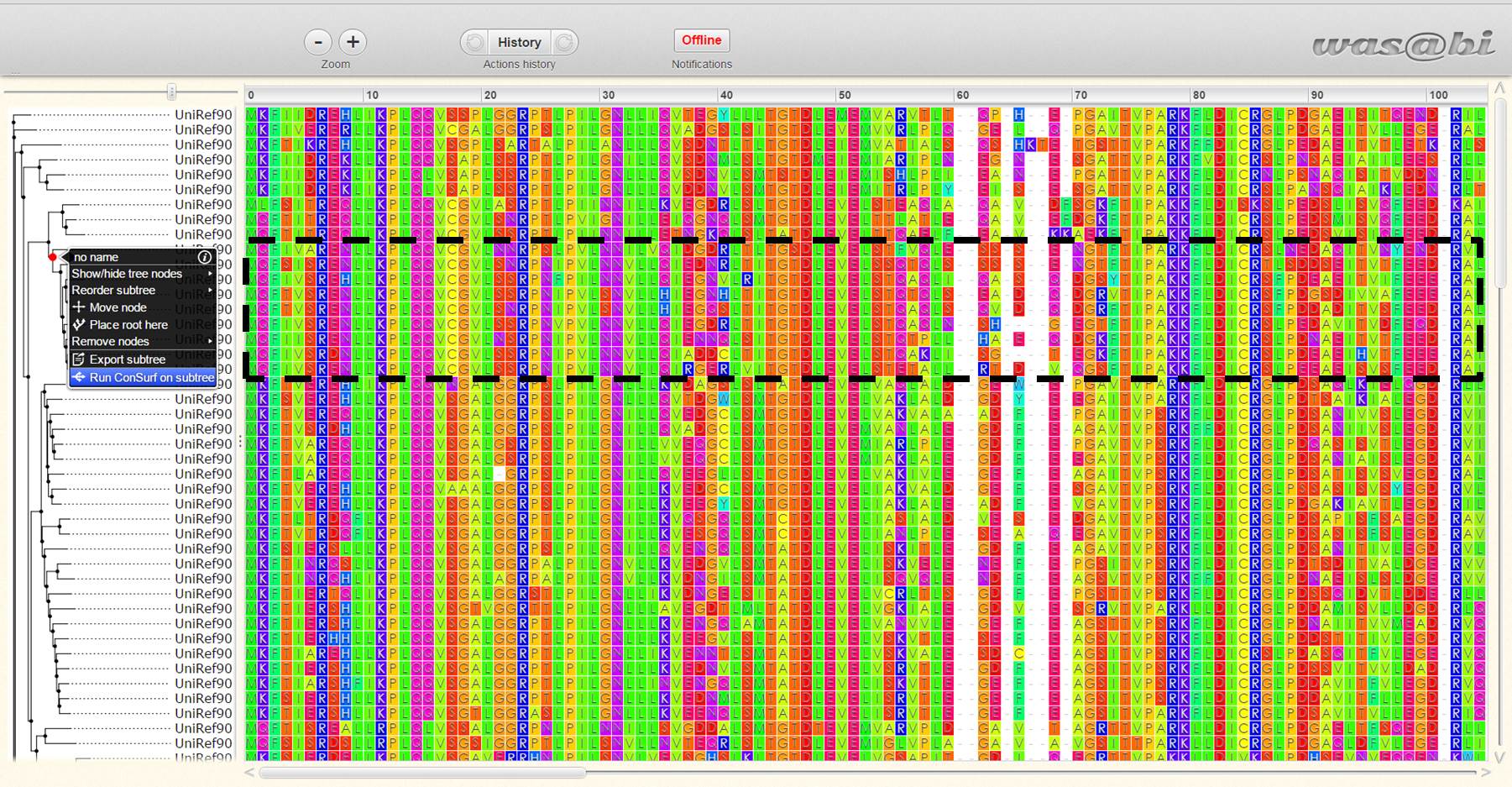
Figure 3: Screenshot of WASABI view of the MSA and phylogenetic tree on the Consurf web server. The option to select sub tree and use it for a new ConSurf run is demonstrated. The subtree is marked by the red circle, and the selected sequences are highlighted with the black rectangle.
Example
As an example we provide the main output of a ConSurf run for the N-terminal region of the GAL4 transcription factor in yeast (PDB ID: 3COQ, chain A and B) in complex with its DNA recognition site( Figure 4). The analysis revealed, as expected, that the functional regions of this protein are highly conserved. For example, all the cysteines that form the Zn(2)-C6 DNA binding domain (CYS11, CYS14, CYS21, CYS28, CYS31, CYS38;(Pan and Coleman, 1990)) were assigned the highest conservation scores. Likewise, PRO26, which is known to be central for DNA binding (Johnston, 1987) is also highly conserved according to our analysis. In addition, other amino acid residues, which are in contact with the DNA (i.e. GLN9, LYS17, LYS18, LYS20, ARG15, LYS23; (Marmorstein et al., 1992)) are relatively conserved. ConSurf was also applied to nucleic acid sequences from yeast, which are the known binding sites of GAL4 and their adjacent neighborhood (Figure 4). As anticipated, the analysis revealed that the consensus pattern CGG-N11-CCG typical to GAL4 binding site is highly conserved. An extended full ConSurf analysis of this example is available in the 'GALLERY' section the ConSurf web site.
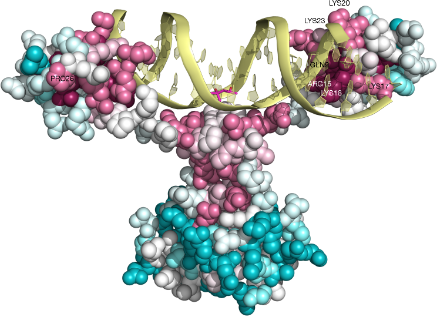
Figure 4: A ConSurf analysis for the GAL4 transcription factor and its DNA binding site. The 3D structure of the N-terminal region of the GAL4 transcription factor in yeast bound to the DNA is presented using a space-filled model. The amino-acids and the nucleotides are colored by their conservation grades using the color-coding bar, with turquoise-through-maroon indicating variable-through-conserved. Positions, for which the inferred conservation level was assigned with low confidence, are marked with light yellow. The figure reveals that the functionally important regions on both the DNA and the protein are highly conserved. The run was carried out using PDB code 3COQ and the figure was generated using the PyMol (DeLano, 2008) script output by ConSurf.
ConSurf vs estimates of the evolutionary conservation without explicit use of phylogenetic relationships
We conducted simulations to examine the accuracy of ConSurf's estimate of the evolutionary rates of the amino acid sites in comparison to entropy-based methods of estimating conservation/divergence, which do not take explicitly into account the phylogenetic relationships among the homologues. We synthesized a benchmark set of multiple sequence alignment, prepared using preset evolutionary rates, and examined the capacity of Rate4Site (Mayrose et al., 2004), the algorithm used in ConSurf, to reproduce these rates. In comparison, we examined the accuracy or reconstructing the rates using entropy based methods of estimating the evolutionary conservation, implemented by (Capra and Singh, 2007).
We compiled a set of multiple sequence alignments (MSAs) of homologous proteins by simulating the evolutionary process using INDELible (Fletcher and Yang, 2009) and preset evolutionary trees. Our set included 25 replicas of 16, 32, 64, 128, 256, and 512 taxa, i.e., a total of 150 trees. These ultrametric trees were generated using Mesquite (Maddison and Maddison, 2015), following a birth-death process with the default birth rate of 0.3 and death rate of 0.1. The tree height of each of these trees was then adjusted to be 0.6. Protein MSA was simulated for each phylogenetic tree by INDELible, using the following parameters: with amino-acid replacements following the LG matrix; Power law insertion/deletion length distribution (a=1.7) with maximum indel length of 30 amino acids; insertion rate which equals to deletion rate and equals to 0.01 relative to average substitution rate of 1; evolutionary rates for each position were drawn from 16 categories discrete gamma distribution with alpha=0.3; The length of the root sequence was set to be 500 amino acids. Overall, these parameters resulted in MSAs which looks biologically reasonable based on visual comparison of the alignments' total length, number and length of indels. The average length of the simulated MSAs were 567.32, 591.92, 663.08, 776.8, 984.64, 1295.92 amino acids for the 16, 32, 64 and 128, 256 and 512 taxa datasets, respectively. In addition we simulated sequences with rate equals to the Shannon entropy scores calculated for the default ConSurf MSA generated for the influenza neuraminidase (PDB ID: 2HU4 chain A) and using phylogenetic tree inferred from the MSA using RAxML. The simulated dataset and all calculated results can be downloaded here.
Each simulated MSA was next provided as input for each of the conservation inference methodologies. It is noteworthy that in rate4site, the phylogenetic tree was inferred from the MSA (rather than using the tree that was used in the simulations). Spearman correlation between the true evolutionary rate used during the simulation of each position and the inferred conservation score was calculated for each of the methods. The results are shown in table 1.
The data in the table 1 shows that Rate4Site is far superior to all examined alternatives; the Spearman correlations between the true (simulated) rates and those inferred by Rate4Site are significantly higher than these of the other entropy based methods. The last row in the table includes comparison to rates derived from a set of highly similar sequences (the influenza neuraminidase), as a particularly difficult test case. Rate4Site is slightly more accurate than the alternatives also in this case.
Table1: Average Spearman correlation coefficients of different conservation methodsand the true evolutionary rate.
|
Number of sequences |
rate4site |
Shannon entropy |
Property entropy |
Property relative entropy |
Relative entropy |
JS divergence |
Sum of pairs |
|
16 |
0.831 (±0.03) |
-0.713 (±0.037) |
-0.614 (±0.045) |
-0.538 (±0.048) |
-0.674 (±0.041) |
-0.690 (±0.039) |
-0.669 (±0.039) |
|
32 |
0.879 (±0.024) |
-0.742 (±0.039) |
-0.652 (±0.046) |
-0.588 (±0.053) |
-0.711 (±0.054) |
-0.733 (±0.042) |
-0.688 (±0.048) |
|
64 |
0.923 (±0.012) |
-0.765 (±0.022) |
-0.686 (±0.027) |
-0.624 (±0.035) |
-0.735 (±0.022) |
-0.763 (±0.02) |
-0.709 (±0.027) |
|
128 |
0.944 (±0.01) |
-0.773 (±0.025) |
-0.700 (±0.03) |
-0.639 (±0.042) |
-0.744 (±0.029) |
-0.772 (±0.026) |
-0.718 (±0.037) |
|
256 |
0.961 (±0.007) |
-0.763 (±0.022) |
-0.697 (±0.027) |
-0.628 (±0.029) |
-0.737 (±0.021) |
-0.768 (±0.018) |
-0.705 (±0.026) |
|
512 |
0.974 (±0.005) |
-0.756 (±0.014) |
-0.692 (±0.022) |
-0.62 (±0.029) |
-0.724 (±0.021) |
-0.757 (±0.016) |
-0.694 (±0.026) |
|
Neuraminidase (134 sequences) |
0.597 (±0.032) |
-0.560 (±0.052) |
-0.448 (±0.054) |
-0.375 (±0.063) |
-0.486 (±0.054) |
-0.559 (±0.053) |
-0.423 (±0.056) |
� For details about entropy based methods see (Capra and Singh, 2007); For Rate4Site details see (Mayrose et al., 2004).
References
- Adachi,J. et al. (2000) Plastid genome phylogeny and a model of amino acid substitution for proteins encoded by chloroplast DNA. J. Mol. Evol., 50, 348-358.
- Adachi,J. and Hasegawa,M. (1996) Model of amino acid substitution in proteins encoded by mitochondrial DNA. J. Mol. Evol., 42, 459-468.
- Akaike,H. (1974) A new look at the statistical model identification.IEEE Trans. Automat. Contr., 19, 716-723.
- Altschul,S.F. et al. (1990) Basic local alignment search tool. J. Mol. Biol., 215, 403-410.
- Altschul,S.F. et al. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res., 25, 3389-402.
- Ashkenazy,H. et al. (2010) ConSurf 2010: calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res., 38, W529-533.
- Ashkenazy,H. et al. (2016) ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules.submitted
- Biegert,A. and Söding,J. (2009) Sequence context-specific profiles for homology searching. Proc. Natl. Acad. Sci. U. S. A., 106, 3770-3775.
- Capra,J.A. and Singh,M. (2007) Predicting functionally important residues from sequence conservation. Bioinformatics, 23, 1875-82.
- Celniker,G. et al. (2013) ConSurf: using evolutionary data to raise testable hypotheses about protein function. Isr. J. Chem., 53, 199-206.
- Darriba,D. et al. (2012) jModelTest 2: more models, new heuristics and parallel computing. Nat. Methods, 9, 772.
- Darriba,D. et al. (2011) ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics, 27, 1164-1165.
- DeLano,W.L. (2008) The PyMOL Molecular Graphics System.
- Finn R. D. et al. (2011) HMMER Web Server: Interactive Sequence Similarity Searching. Nucleic Acids Res. 39, W29-37.
- Fletcher,W. and Yang,Z. (2009) INDELible: a flexible simulator of biological sequence evolution. Mol. Biol. Evol., 26, 1879-88.
- Glaser,F. et al. (2003) ConSurf: identification of functional regions in proteins by surface-mapping of phylogenetic information. Bioinformatics, 19, 163-164.
- Hasegawa,M.et al. (1985) Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J. Mol. Evol., 22, 160-174.
- Herráez,A. (2006) Biomolecules in the computer: Jmol to the rescue. Biochem. Mol. Biol. Educ., 34, 255-61.
- Johnston,M. (1987) Genetic evidence that zinc is an essential co-factor in the DNA binding domain of GAL4 protein. Nature, 328, 353-5.
- Jones,D.T. et al. (1992) The rapid generation of mutation data matrices from protein sequences. Bioinformatics, 8, 275-282.
- Jukes,T.H. and Cantor,C.R. (1969) Evolution of protein molecules. In, Munro,H.N. (ed), Mammalian Protein Metabolism, pp. 21-132.
- Landau,M. et al. (2005) ConSurf 2005: the projection of evolutionary conservation scores of residues on protein structures. Nucleic Acids Res., 33, W299-302.
- Le,S.Q. and Gascuel,O. (2008) An improved general amino acid replacement matrix. Mol. Biol. Evol., 25, 1307-1320.
- Li,W. and Godzik,A. (2006) Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics, 22, 1658-1659.
- Lorenz,R. et al. (2011) ViennaRNA Package 2.0. Algorithms Mol. Biol., 6, 26. M.O. Dayhoff, R.M. Schwartz, and B. C,O. (1978) A model of evolutionary change in proteins. Maddison,W.P. and Maddison,D.R. (2015) Mesquite: a modular system for evolutionary analysis. Version 3.04. http://mesquiteproject.org.
- Marmorstein,R. et al. (1992) DNA recognition by GAL4: structure of a protein-DNA complex. Nature, 356, 408-14.
- Mathews,D.H. et al. (2004) Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA secondary structure. Proc. Natl. Acad. Sci. U. S. A., 101, 7287-7292.
- Mayrose,I. et al. (2004) Comparison of site-specific rate-inference methods for protein sequences: empirical Bayesian methods are superior. Mol. Biol. Evol., 21, 1781-
- Meier,A. and Söding,J. (2015) Automatic prediction of protein 3D structures by probabilistic multi-template homology modeling. PLoS Comput. Biol., 11, e1004343.
- Pan,T. and Coleman,J.E. (1990) GAL4 transcription factor is not a 'zinc finger' but forms a Zn(II)2Cys6 binuclear cluster. Proc. Natl. Acad. Sci. U. S. A., 87, 2077-81.
- Pettersen,E.F. et al. (2004) UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem., 25, 1605-12.
- Pupko,T. et al. (2002) Rate4Site: an algorithmic tool for the identification of functional regions in proteins by surface mapping of evolutionary determinants within their homologues. Bioinformatics, 18 Suppl 1, S71-77.
- Remmert,M. et al. (2012) HHblits: lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat. Methods, 9, 173-175.
- Rose,P.W. et al. (2015) The RCSB protein data bank: views of structural biology for basic and applied research and education. Nucleic Acids Res., 43, D345-356.
- Rost,B. (1999) Twilight zone of protein sequence alignments. Protein Eng., 12, 85-94.
- Saitou,N. and Nei,M. (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol., 4, 406-25.
- Sali,A. and Blundell,T.L. (1993) Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol., 234, 779-815.
- Sayle,R.A. and Milner-White,E.J. (1995) RASMOL: biomolecular graphics for all. Trends Biochem. Sci., 20, 374.
- Steinegger, M. and Söding, J. (2017) MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nature Biotechnology. 35: 1026-1028.
- Susko,E. et al. (2002) Testing for differences in rates-across-sites distributions in phylogenetic subtrees. Mol. Biol. Evol., 19, 1514-23.
- Tamura,K. (1992) Estimation of the number of nucleotide substitutions when there are strong transition-transversion and G+C-content biases. Mol.Biol. Evol., 9, 678-
- Tavare,S. (1986) Some probabilistic and statisical problems on the analysis of DNA sequences. Lect. Math. Life Sci., 17, 57-86.
- Thornton,J.M. et al. (2000) From structure to function: approaches and limitations. Nat. Struct. Biol., 7 Suppl, 991-4.
- Veidenberg,A. et al. (2015) Wasabi: an integrated platform for evolutionary sequence analysis and data visualisation. Mol. Biol. Evol., msv333.
- Whelan,S. and Goldman,N. (2001) A General empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach. Mol. Biol. Evol., 18,691-699.