1. What is the minimum number of sequences required to get reliable results?
2. What if I get only few homologous sequences for my query protein?
3. What are the advantages of using phylogenetic tree?
4. Should I use the Bayesian or the Maximum Likelihood method?
6. What should I do if I find problems uploading external multiple sequence alignment (MSA) files?
7. How can I produce a ConSurf picture?
8. Are there pre-calculated conservation grades available? What is ConSurf-DB?
9. What if I do not have a structure?
10. How reliable are the conservation scores?
12. What is the 'Clean_Uniprot' database?
-
What is the minimum number of sequences required to get reliable results?
There is no exact answer to this question, as the sequence variability matters. Nevertheless, as a 'rule of thumb' we recommend a minimum of 10 homologues. If PSI-BLAST found fewer than that, you can try to raise the E-value cutoff, for example by changing it from the default 0.001 to 0.01. However, the additional sequences found may be phylogenetically distant from the query sequence, which can influence the quality of the generated multiple sequence alignment. On the other hand, if remote homologues are added, the conservation signal/s may decrease due to background noise added to the multiple sequence alignment. You can control the number and diversity of the input sequences by selecting the relevant sequences out of BLAST results (check the box titled 'Let me select the sequences for the analysis manually out of Blast results') or by providing your own alignment file.
-
What if I get only few homologous sequences for my query protein?
One of the issues in retrieving homologous sequences is choosing the database used for the search. For example, certain organisms are mostly represented in the trEMBL database but not in SwissProt. Few databases can be considered in such cases:
- The Uniprot database contains sequences from both SwissProt and trEMBL, thus a possible solution is to try running ConSurf with the Uniprot database.
- It is highly recomended to use Clean_UniProt which is a modified version of the UniProt database aimed to screen the more reliable sequences based on two criteria: (i) if the "Decription" (DE) field contain "Disease", "RIKEN", "variant", "mutation", "mutant" or "whole genome shotgun sequence" the sequence is removed; (ii) if the database is "TrEMBL" and the "Comments" (CC) lines contain the word "CAUTION" the sequence is removed.
- The NCBI nr database comprised of all non-redundant GenBank CDS translations, PDB, SwissProt, PIR and PRF contains more sequences than Uniprot and is another option that should be considered
Another option is to raise the "PSI-BLAST E-value Cutoff" parameter. The results will be less reliable thus should be more carefully examined.
-
What are the advantages of using phylogenetic tree?
- All databases contain a certain degree of over representation of certain families or species (HIV, Human, etc.). Phylogenetic trees deal better with redundancy than methods that analyze multiple sequence alignment directly, by weighting clusters of closely related sequences differently. This clustering process diminishes the influence of redundant sequences.
- The phylogenetic tree together with the model of sequence evolution describe the evolutionary processes that generated the multiple sequence alignment. By taking the tree explicitly into account when computing conservation scores, it is possible to better identify the amino-acid replacements that could have occurred in the history of a family of homologous sequences, thus increasing the accuracy of the calculation.
- The phylogenetic tree is a prerequisite for using probabilistic based approaches such as the maximum-likelihood and the Bayesian approach. The branch lengths of the tree, for example, are needed for computing the probabilities of amino-acid replacements. These statistically reliable methods increase the statistical reliability of the calculations and allow us to compute confidence intervals around the estimated conservation scores in the Bayesian method.
-
Should I use the Bayesian or the Maximum Likelihood method?
The Bayesian method was shown to significantly improve the accuracy of conservation scores estimations over the Maximum Likelihood method. This is particularly important when a small number of sequences are used for the calculations. An additional advantage of the Bayesian method is that a confidence interval is assigned to each of the inferred evolutionary conservation score. A detailed description of the Bayesian methodology is provided in Mol. Biol. Evol., 21, 1781-1791; 2004, (PDF). A detailed description of the Maximum Likelihood methodology is provided in Bioinformatics, 18, S71-77; 2002 (PDF). Rate4Site, a stand-alone application that implements both algorithms, is available at the URL: Rate4Site. We recommend running the target protein using both methods, which will provide a double check for your results.
-
Is it possible for the extreme grades 1 and 9 to be unoccupied? Which conditions give this result?
Grades 1-8 can be unoccupied, although this will occur rarely, such as when ConSurf finds few homologues. Grade 9 is always occupied by at least one residue.
-
What should I do if I find problems uploading external multiple sequence alignment (MSA) files?
- Check your MSA file in a simple text editor (e.g. Notepad on Windows). It is very common that MSA files downloaded from the web contain unnecessary characters. Eliminate them, and save your file as text only.
- Some kind of incompatibility between the text format of PC / Unix and Mac machines exists. If you are running the ConSurf server from a Mac platform, and you get repetitive error messages, we recommend that you save your file using Word as an "MS-Dos" text file. This format should be compatible with the Dos and Unix text files.
- If none of this works, please contact us!!
-
How can I produce a ConSurf picture?
- Using 'Chimera' follow the instructuions under the link titled 'Follow the instructions to produce a Chimera figure (For users of Chimera)' in your output results page
- Using 'PyMol' follow the instructions under the link titled 'Follow the instructions to produce a PyMol figure (For users of PyMol)' in your output results page
-
To produce a final high-quality picture we recommend one the "spheres" or "surface" representation:
"pymol>>show spheres, xxx"
"pymol>>show surface, xxx"
and then of course 'ray' and save the picture:
"pymol>>ray"
"pymol>>png xxx.png"
- Create high resolution images and animations using PolyView-3D server (see help).
- Copy the image directly. Here are instructions for grabbing a snapshot directly from the molecular image on your screen.
-
When publishing figures from ConSurf, we recommend including our visual color key:
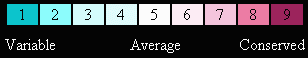
and here is our color-blind friendly color key:
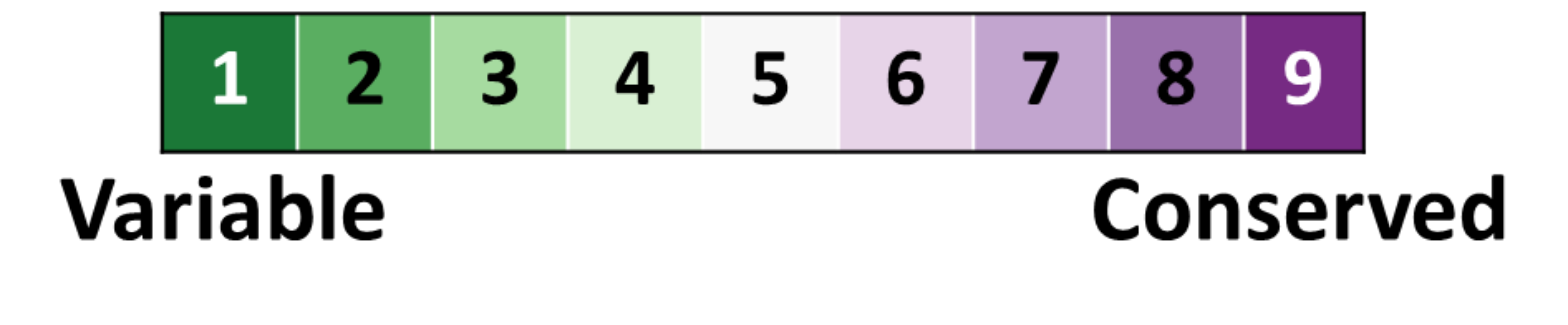
Right click on the above color key image to save it to your disk.
Here is an example of a figure that includes the color key: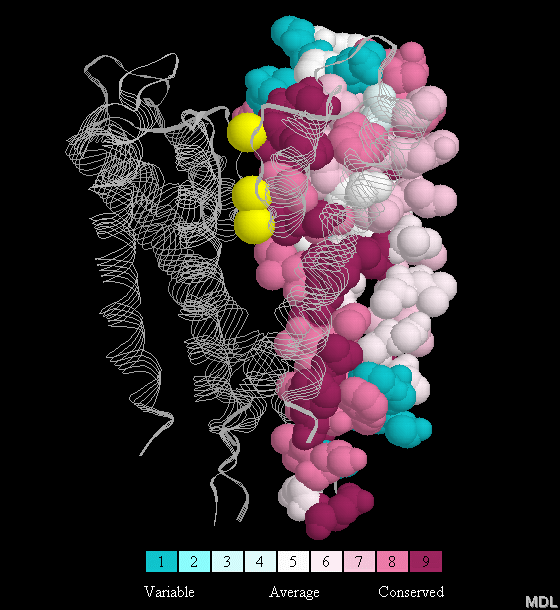
-
Are there pre-calculated conservation grades available? What is ConSurf-DB?
The ConSurf-DB database provides pre-calculated results. A description of the server can be found in http://consurfdb.tau.ac.il/ and in the paper "The ConSurf-DB: Pre-calculated evolutionary conservation profiles of protein structures" Nucleic Acids Research, 2009, Vol. 37, Database issue D323-D327 (PDF) (Online version).
-
What if I do not have a structure?
You can use ConSurf with sequence only (nucleotides/amino-acids). For protein sequence you can also observe ConSurf results projected on structures of homologues proteins (if available). Alternatively, you can run ConSurf with a model structure.
-
How reliable are the conservation scores?
The computation of conservation scores was tested many times. Basically, we have tested it using simulations:
Mayrose, I., Graur, D., Ben-Tal, N., and Pupko, T. 2004. Comparison of site-specific rate-inference methods: Bayesian methods are superior. Mol. Biol. Evol. 21(9): 1781-1791. PDF
The rates seem to converge to the true value when the number of sequences increases.
We also evaluated the impact of a wrong tree topology on rate estimates: Mayrose, I., Mitchell, A., and Pupko, T. 2005. Site-specific evolutionary rate inference: taking phylogenetic uncertainty into account. J. Mol. Evol. 60(3):345-353. PDF
The rate inferred by rate4site algorithm are also usually highly correlated with a Ka/Ks score that is computed based on the coding DNA sequences.
Finally but maybe most important, it seems to give biology reasonable results.
On the other hand, you can never be sure that the code (which is huge in rate4site) has no bugs. -
What is the best way to collect homologous sequences in order to construct an MSA?
Which protein databases should be used?
How does ConSurf do it?The answer to this question is not straightforward; it depends on the information we want to obtain from the multiple sequence alignment (MSA). If you know your protein well, in ConSurf you can choose the relevant sequences for the analysis manually out of BLAST results. For a general conservation analysis of a certain protein family/superfamily, where the aim is to point out the major active site or structural elements that are important for determination of the overall fold, it is advised to collect as many homologues as possible. In contrast, when searching for a functional site that is specific for a certain family/sub-family, the alignment should be smaller and include only close homologues that share the function. The caveat is that, in reality, we might not know which of the proteins share the same function.
Generally, there are two kinds of homologues to collect:
- Orthologs: namely, the same protein from different species. These proteins conduct the exact same function and therefore share a high sequence similarity. Orthologs should be used for analysis of specific functions that are shared by a protein family/sub-family.
- Paralogs: that is, homologous proteins that can be found within the same species. These proteins diverged from a common ancestor but their function may have changed to some degree during evolution. Thus, they share lower sequence similarity, and should be used to explore the major structural and functional characteristics of the protein family/superfamily.
Our experience has been that the SWISSPROT database is recommended in search for close homologous proteins. It yields highly annotated sequences for small collections of a small amount of species. For a broader analysis (more species and more sequences that are less annotated), the CLEAN_UNIPROT (our filterd UNIPROT database) database should be used.
The choice of the database should also be determined by the query protein. If it belongs to a large family such as the kinases or proteases, SWISSPROT, which provides hundreds of sequences with a low BLAST E-value, is sufficient. If it is a less abundant protein, CLEAN_UNIPROT or UNIPROT is recommended.
The issue of redundancy is not crucial when using ConSurf, since the algorithm that calculates the conservation scores takes the phylogenetic relationships within the alignment into account. This is actually one of the strongest qualities of ConSurf. It means that the conservation of positions of proteins in species that are more "evolutionary distant" is more significant and vice-versa. For example, a position conserved between human and chimpanzee, is not necessarily noteworthy, since the two species diverged recently. On the other hand, if the position is conserved in human and in flies or worms, then it may indicate importance for structure or function. Nevertheless, if a certain family is over-represented in the alignment (namely, it has more orthologs in the database) it could have an effect on the construction of the alignment, but it is not very crucial.
However there are cases in which all the top sequences found by searching SWISS-PROT/UNIPROT share high similarity between each other and do not contain sufficient data to estimate the evolutionarily conservation. In such cases it is recommended to search against the UniRef90 DB where the redundancy is removed on the database level and thus to find more relevant sequences. To remove redundancy one may use ConSurf option to specify redundancy level for which sequences are clustered and redundancy is removed (it is recommended to remove sequences that are more than 85%-90% conserved). This would probably remove most close orthologs, especially when analyzing a protein domain.
Finally, it is important to mention that there are databases of readymade multiple sequence alignments that one could use. For example: ENSEMBL, PFAM, RFAM, ORTHOMAM. A multiple sequence alignment from one of these databases may be used as is, or taken as a basis to construct a more suitable one, e.g., by the elimination of unwanted sequences. -
What is the 'Clean_Uniprot' database?
'Clean_Uniprot' is a modified version of the UniProt database aimed to screen the more reliable sequences based on two criteria:
- If the "Decription" (DE) field contain "Disease", "RIKEN", "variant", "mutation", "mutant" or "whole genome shotgun sequence" the sequence is removed;
- If the database is "TrEMBL" and the "Comments" (CC) lines contain the word "CAUTION" the sequence is removed.
-
Why did you recently change the defaults in the procedure for collecting homologous sequences in ConSurf?
The accuracy of the ConSurf conservation scores depends on the number and quality of homologous sequences. When developing the first version of ConSurf [PDF], some 10 years ago, we found that usually 50 homologues from SwissProt give the best results. However, the inflation in the sequence databases since then made us recommend a new default procedure. We now suggest using UniRef90, which contains man more sequences and removes redundancy at a level of 90% sequence identity. We also increased the default number of putative homologous sequences to 150, but reduced the PSI-BLAST E-value cutoff to 0.0001 to minimize the risk of including non-homologues.